


 Most CYP450 enzymes are also inducible.11 Drugs or endogenous compounds bind to a transcription factor, which stimulates an increased synthesis of the enzyme, thus leading to increased enzymatic activity (Figure 1.4). The
transcription of other proteins associated with drug handling, such as transporter proteins, may also be induced by these perpetrator agents.
Most CYP450 enzymes are also inducible.11 Drugs or endogenous compounds bind to a transcription factor, which stimulates an increased synthesis of the enzyme, thus leading to increased enzymatic activity (Figure 1.4). The
transcription of other proteins associated with drug handling, such as transporter proteins, may also be induced by these perpetrator agents. 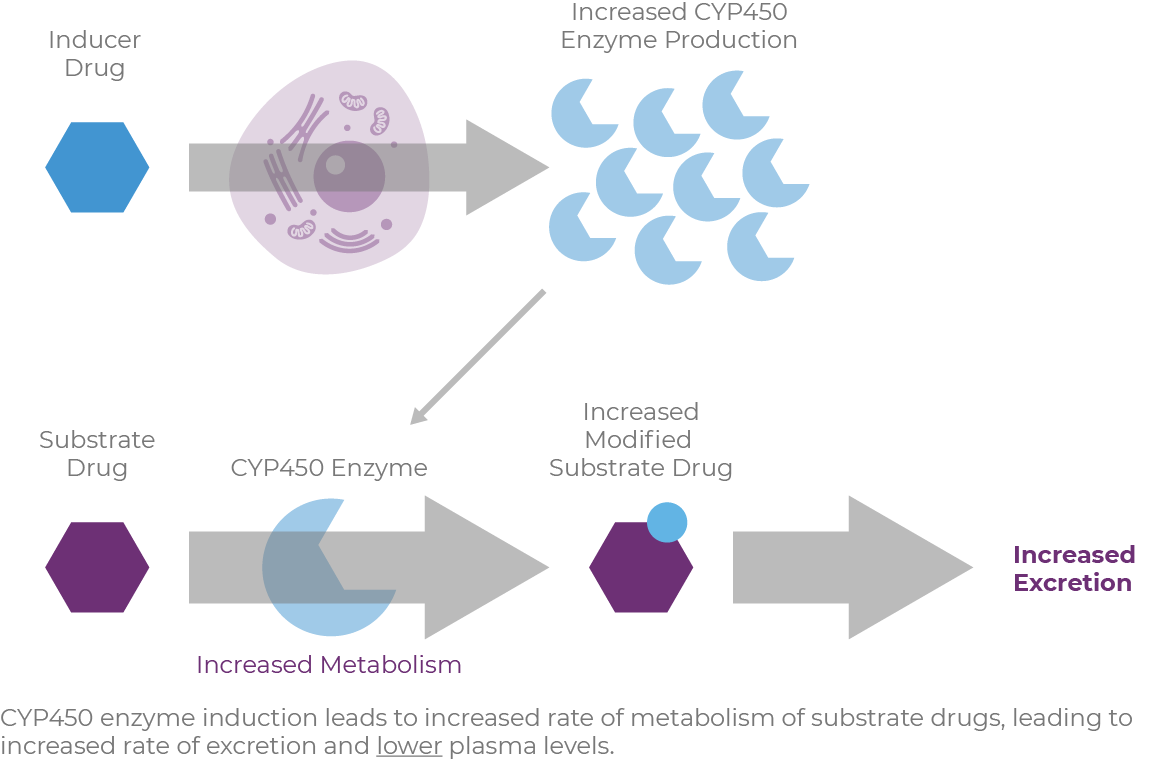
The CYP450 enzymes also display genetic diversity, which can affect their rate of metabolizing activity.12 In this case, the amount of enzyme stays constant, but the activity level is lower or higher.9 A patient with diminished activity is described as a "poor metabolizer," and those with excessive activity are called "ultra" or "rapid metabolizers."
CYP3A4/5 and CYP2D6 are the three most common metabolic pathways within the CYP450 enzyme family, with 50% of drugs being metabolized through CYP3A4/5 pathways and 25% through CYP2D6.13
CYP3A4 is the most predominant metabolic pathway of the commonly prescribed medications,7 and it is highly expressed in the liver and intestine.11 Induction of the CYP3A4 enzyme increases the rate of metabolism of victim compounds, causing lower plasma drug levels.14 This may result in subtherapeutic levels of drugs, or supratherapeutic levels of a drug administered as a prodrug.12 Inducers include phenobarbital, phenytoin, St. John's Wort, and glucocorticoids.15
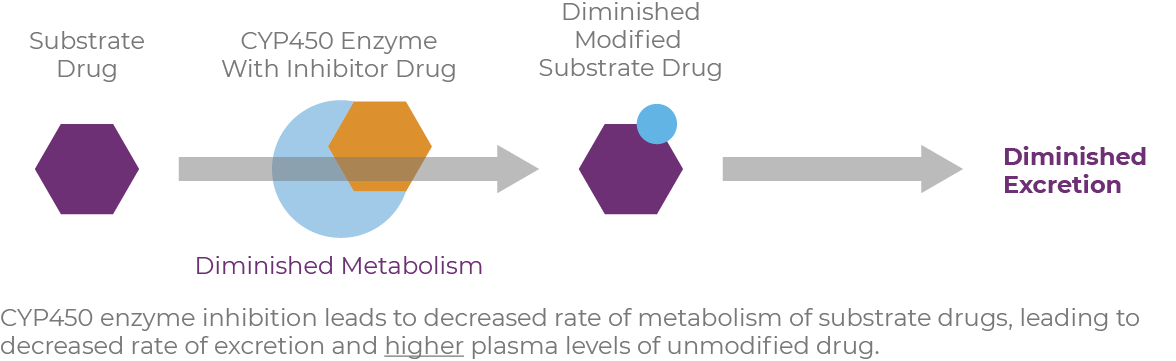
CYP3A4 inhibitors include macrolide antibiotics, azole antifungals, calcium-channel blockers, goldenseal, and grapefruit.15 Inhibition by these agents can result in higher levels of the substrate compound, and, in the case of a prodrug, lower levels of active compound are produced while the enzyme activity is inhibited.12,14
CYP2D6 is another enzyme commonly implicated in drug metabolism.9 CYP2D6 also shows broad substrate specificity but is not highly inducible. CYP2D6 is subject to inhibition by various drugs, including some opiate analogs and some selective serotonin reuptake inhibitors.9,16 CYP2D6 exhibits high genetic variability, which modifies the level of synthesis and the level of enzymatic activity, which can affect individual patients' responses.9 With its wide genetic variation, the degree of enzyme activity can vary greatly between patients, but is usually described in terms of patients being poor metabolizers, intermediate metabolizers, extensive metabolizers, and ultrarapid metabolizers.16
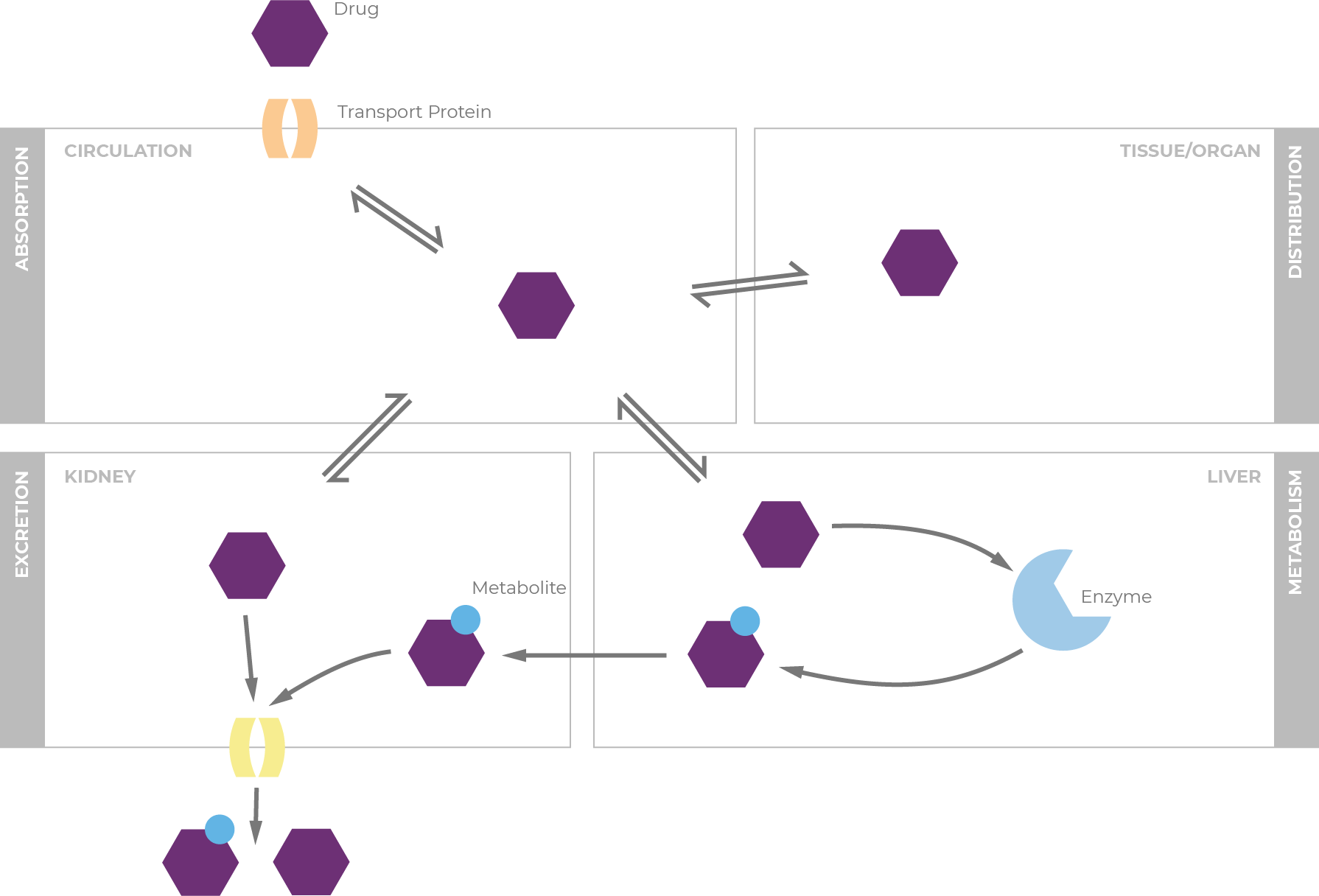
Transporters are membrane-bound proteins that help guide the movement of substrates across biological membranes.17 They are widely distributed in the body, including in the liver, intestine, kidney, and organs with barrier functions. Transporter proteins affect absorption, distribution, and elimination of drugs throughout the body, and altered transporter activity also affects plasma levels of drugs.
As with CYP450 enzyme pathways, a drug may be a substrate of one or many transport proteins, and can be an inducer or inhibitor of transport proteins.7 P-glycoprotein (P-gp) is a well-known efflux transporter. Transport proteins are regulated in similar way to CYP450 enzymes: through constitutive expression and by induction via exogenous compounds.18 In fact, P-gp and CYP3A4 share the same induction pathways, and transporter proteins can also be inhibited through interaction with a perpetrator molecule. Contributions of P-gp competition/induction on DDIs, therefore, are difficult to untangle from processes involved in other metabolic DDIs because the transport proteins and CYP enzymes are frequently co-affected.19
A recent study determined that transporter information is unavailable for 58% of the drugs approved between 2005 and 2016, and for 69% of drugs already established.7 Studies have shown, however, that aripiprazole, paliperidone, and risperidone are substrates of P-gp.20


An estimated 43 million Americans suffer from schizophrenia, bipolar disorder, and/or major depressive disorder, for which APDs are standard of care.1-7 While APDs help manage these disorders, their use is associated with the development of tardive dyskinesia (TD), a condition characterized by involuntary movements of the orofacial region, trunk, and extremities, and which are often irreversible.8,9 Discontinuing or reducing the APD dose to reduce or eliminate TD movements is often not an option in psychiatric patients, as doing so can compromise the patient's underlying mental health condition.10,11
The TD patient population is diverse; patients with TD may have an underlying mental health disorder, and they are also likely to have comorbid conditions, including diabetes, obesity, and cardiovascular and lipid disorders, which require additional medications for management.5-7,12 As such, polypharmacy is especially prevalent in this patient population (Table 2.1).13 In fact, in one retrospective review more than 50% of patients visiting outpatient psychiatry departments were reported to be taking three or more psychotropic drugs, and more than half of patients (53%) reported also taking medications for nonpsychiatric conditions.14 Polypharmacy is associated with several concerns, including the increased risk of AEs, drug interactions, medication error, patient nonadherence, and medical comorbidities. Therefore, treatment considerations for TD must take into account the patients' concomitant medications for psychiatric and nonpsychiatric conditions, to help reduce the risk of DDIs.15,16
| • | Aged >62 years |
| • | Cognitive impairment |
| • | Developmental disability |
| • | Frailty |
| • | Lack of a primary care physician |
| • | Mental health conditions |
| • | Multiple chronic conditions |
| • | Residing in a long-term care facility |
| • | Seeing multiple subspecialists |
Overall, approximately 20% to 30% of adverse reactions are the result of DDIs, and the risk for DDIs increases to about 84% when the number of concomitant drugs rises from 2 to 6 agents.16 Therefore, identifying the metabolic pathways involved in the absorption, metabolism, and excretion of each medicine that a patient is taking is essential to minimize DDIs and adverse reactions.
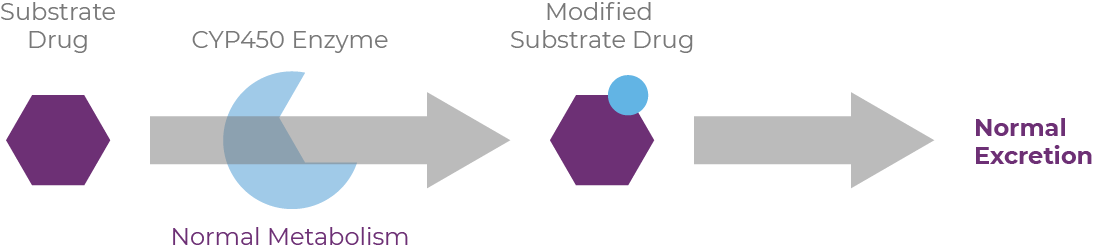
Drugs that serve as substrates, inhibitors, or inducers can have pharmacokinetic interactions with other medications, as well as with foods, beverages, and herbal supplements.16 When drug substrates are metabolized through the cytochrome P450 (CYP450) pathway, CYP inducers cause increased enzyme activity, resulting in reduced drug activity.17 Conversely, CYP inhibitors simultaneously block activity, causing increased plasma levels of drugs or other substrates, sometimes necessitating dosage reductions.
When reviewing patients' medications, it's important to know how each medication will affect the body's handling of concomitant drugs. A so-called "perpetrator" inhibits or induces metabolic enzymes or transporters that affect the plasma levels of a substrate drug, termed the "victim."18 Many inducers and inhibitors are also substrates of the enzymes they affect.19
In addition, the majority of the CYP genes are polymorphic; variation in CYP content and activities can substantially impact the metabolism of drugs, resulting in differences in drug response and adverse drug effects.20
| • | Antiarrhythmics |
| • | Antibiotics |
| • | Antidepressants |
| • | Antifungals |
| • | Antihypertensives |
| • | Antiretrovirals/antivirals |
| • | Antibiotics |
| • | Anticonvulsants/sedatives |
| • | Antifungals |
| • | Antihypertensives |
| • | Corticosteroids |
| • | Wakefulness-promoting agents |
| • | Antianginals |
| • | Antidiabetics |
| • | Antihistamines |
| • | Antihypertensives |
| • | Corticosteroids |
| • | Diuretics |
| • | Statins |
| • | Vasodilators |
| • | Antiarrhythmics |
| • | Antidepressants |
| • | Antifungals |
| • | Antihistamines |
| • | Antiparasitics |
| • | Antipsychotics |
| • | Calcium reducers |
| Evidence suggests that unlike most other CYP450 enzymes, CYP2D6 is not very susceptible to enzyme induction |
| • | Antiarrhythmics |
| • | Antiemetics |
| • | Antihistamines |
| • | Antihypertensives |
| • | Antidepressants |
| • | Antipsychotics |
The CYP3A4/5 pathway metabolizes many medications used to manage components of metabolic syndrome, such as antihypertensives, antidiabetics, and statins, which could interact with APDs or medicines used to treat TD (Table 2.2).21-24 Some of the common substrates of the CYP2D6 pathway are beta blockers, APDs, antiarrhythmics, antiemetics, antidepressants, and opioids (Table 2.3).20,21,23,24,26
To minimize or prevent DDIs in patients receiving APD therapy, clinicians should identify concomitant medications that are metabolized by CYP3A4/5 or CYP2D6 (see Tables 2.2 and 2.3), those with a narrow therapeutic index, or those requiring stable serum concentrations, such as anticoagulants, anticonvulsants, anti-infectives, cardiovascular therapeutics, oral contraceptives, glucocorticoids, antiretroviral agents, and other psychotropics.16
Membrane-bound proteins called transporters also play a role in the metabolism of drugs, although less is known about their role in DDIs.27,28 Found in the liver, intestine, kidney, and organs with barrier functions, transporter proteins enable the translocation of substrate molecules across the biological membranes.27 In doing so, they affect absorption, distribution, and drug elimination.29 Breast cancer resistance protein (BCRP) and P-glycoprotein (P-gp)/multidrug resistance-associated protein (MDR)-1 are well known efflux transporters, and their inhibition results in decreased efflux of substrate drugs; further, their induction leads to increased efflux of substrate drugs.28 Many drugs that are CYP3A4 substrates, inhibitors, and inducers are also inhibitors or inducers of P-gp.30 As such, many drug interactions involve effects of both CYP3A4 and P-gp.
Managing TD in patients taking concomitant medications for other conditions requires that clinicians are aware of all medications a patient is taking, for both psychiatric and nonpsychiatric conditions, as these can have important safety and therapeutic consequences, as well as can affect adherence.16
Consideration of DDIs is especially important in elderly patients, as this population has a significantly higher risk for TD and is often polymedicated.5,33 Furthermore, elderly patients are more vulnerable to certain AEs (eg, orthostatic hypertension, urinary retention, and constipation), which may require additional medications for management.5 Managing these AEs with new medications, in turn, may compound the risk of additional AEs and DDIs.
The 2021 American Psychiatric Association practice guidelines recommend the use of a vesicular monoamine transporter 2 (VMAT2) inhibitor for the treatment of TD that has an impact on the patient.34 VMAT2 inhibitors are the only FDA-approved drugs for the treatment of TD in adults.31,35 When choosing a VMAT2 inhibitor, it is important to consider the patient's overall treatment plan, and to identify drug metabolic pathways that may be involved in DDIs.
1. United States Census Bureau, Population Division. Annual estimates of the resident population: April 1, 2010 to July 1, 2017 . Accessed March 31, 2023. https://www2.census.gov/programs-surveys/popest/tables/2010-2017/national/totals/PEPANNRES.pdf 2. National Institute of Mental Health. Schizophrenia. NIMH website. Updated May 2018. Accessed May 11, 2021. https://www.nimh.nih.gov/health/statistics/schizophrenia.shtml 3. National Institute of Mental Health. Any mood disorder. NIMH website. Updated November 2017. Accessed May 11, 2021. https://www.nimh.nih.gov/health/statistics/any-mood-disorder.shtml 4. National Institute of Mental Health. Bipolar disorder. NIMH website. Accessed May 13, 2021. https://www.nimh.nih.gov/health/statistics/bipolar-disorder 5. Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(2 suppl):1-56. 6. Hirschfeld RM, Bowden CL, Gitlin MJ, et al. Practice guideline for the treatment of patients with bipolar disorder, second edition. Accessed August 5, 2020. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/bipolar.pdf 7. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. Accessed August 5, 2020. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf 8. Caroff SN, Campbell EC, Carroll B. Pharmacological treatment of tardive dyskinesia: recent developments. Expert Rev Neurother. 2017;17(9):871-881. 9. Kim J, MacMaster E, Schwartz TL. Tardive dyskinesia in patients treated with atypical antipsychotics: case series and brief review of etiologic and treatment considerations. Drugs Context. 2014;3:212259. 10. Caroff SN, Miller DD, Dhopesh V, Campbell EC. Is there a rational management strategy for tardive dyskinesia? Decisions should be based on the course of TD and effective control of psychotic symptoms. Curr Psychiatry. 2011;10(10):22-32. 11. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469. 12. Barnes TR, Bhatti SF, Adroer R, Paton C. Screening for the metabolic side effects of antipsychotic medication: findings of a 6-year quality improvement programme in the UK. BMJ Open. 2015;5(10):e007633. 13. Halli-Tierney AD, Scarbrough C, Carroll D. Polypharmacy: evaluating risks and deprescribing. Am Fam Physician. 2019;100(1):32-38. 14. Weinstock LM, Gaudiano BA, Epstein-Lubow G, Tezanos K, Celis-Dehoyos CE, Miller IW. Medication burden in bipolar disorder: a chart review of patients at psychiatric hospital admission. Psychiatry Res. 2014;216(1):24-30. 15. Caroff SN. Overcoming barriers to effective management of tardive dyskinesia. Neuropsychiatr Dis Treat. 2019;15:785-794. 16. Mora F, Molina JD, Zubillaga E, López-Muñoz F, Álamo C. CYP450 and its implications in the clinical use of antipsychotic drugs. J Clin Exp Pharmacol. 2015;5(3). 17. Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391-396. 18. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions: Guidance for Industry. January 2020. Accessed August 25, 2021. https://www.fda.gov/media/134581/download 19. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34(1-2):17-35. 20. Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48(11):689-723. 21. National Institutes of Health, US National Library of Medicine. MedlinePlus. Drugs, Herbs and Supplements. Published April 28, 2015. Accessed June 11, 2020. https://medlineplus.gov/druginfo/meds 22. Horn JR, Hansten PD. Get to know an enzyme: CYP3A4. Pharmacy Times. September 1, 2008. Accessed April 29, 2021. https://www.pharmacytimes.com/view/2008-09-8687 23. US Food & Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Accessed May 5, 2021. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table5-1 24. PubChem. National Center for Biotechnology Information. PubChem Compound Database. Accessed August 25, 2021. https://pubchem.ncbi.nlm.nih.gov/ 25. Home - PMC - NCBI. National Center for Biotechnology Information. Accessed June 11, 2020. https://www.ncbi.nlm.nih.gov/pmc/articles 26. Horn JR, Hansten PD. Get to know an enzyme: CYP2D6. Pharmacy Times. July 1, 2008. Accessed April 29, 2021. https://www.pharmacytimes.com/view/2008-07-8624 27. Gupta SK, Singh P, Ali V, Verma M. Role of membrane-embedded drug efflux ABC transporters in the cancer chemotherapy. Oncol Rev. 2020;14(448):144-151. 28. Saravanakumar A, Sadighi A, Ryu R, Akhlaghi F. Physicochemical properties, biotransformation, and transport pathways of established and newly approved medications: a systematic review of the top 200 most prescribed drugs vs. the FDA-approved drugs between 2005 and 2016. Clin Pharmacokinet. 2019;58(10):1281-1294. 29. Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26(9):2039-2054. 30. Horn JR, Hansten PD. Drug interactions with CYP3A4: an update. Accessed May 5, 2021. https://www.pharmacytimes.com/view/drug-interactions-with-cyp3a4-an-update 31. Ingrezza® (valbenazine) capsules. Prescribing Information. San Diego, CA: Neurocrine Biosciences, Inc. 32. Citrome L. Valbenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(7):10.1111/ijcp.12964. 33. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425. 34. Keepers GA, Fochtmann LJ, Anzia JM, et al. The American Psychiatric Association practice guideline for the treatment of patients with schizophrenia. Washington, DC: American Psychiatric Association; 2021. 35. AUSTEDO® XR (deutetrabenazine) extended-release tablets/AUSTEDO® current Prescribing Information. Parsippany, NJ: Teva Neuroscience, Inc.
TD is a potentially disabling, often irreversible, movement disorder that results from chronic exposure to typical or atypical antipsychotic drugs (APDs).1-3 TD classically presents as involuntary, repetitive movements throughout the body, most often seen in the mouth, tongue, and jaw.1,2,4 TD of any severity can have a substantial impact on a patient's functioning, quality of life, and underlying psychiatric stability.2,5-10 Therefore, timely recognition of TD movements is imperative for appropriate management and treatment.11
The American Psychiatric Association practice guidelines recommend vesicular monoamine transporter 2 (VMAT2) inhibitors for the treatment of TD that has an impact on the patient.11 Typically, patients with TD have multiple underlying health conditions, requiring a range of medications. In order to limit drug-drug interactions, it is important to consider pharmacokinetics and metabolic pathways when choosing a VMAT2 inhibitor to treat TD.12-14
AUSTEDO XR is a VMAT2 inhibitor indicated for the treatment of TD in adults.14 The precise mechanism of action of AUSTEDO XR is unknown, but it is believed to be related to its effect as a reversible depletor of monoamines, including dopamine, from nerve terminals. AUSTEDO XR inhibits the uptake of dopamine into synaptic vesicles by binding to VMAT2 on the vesicle in the presynaptic neuron.14,15 Dopamine molecules collect outside the blocked VMAT2 and are degraded by monoamine oxidase. Reducing dopamine levels in the presynaptic neuron results in less dopamine signaling to the postsynaptic neuron, which may lead to fewer abnormal involuntary movements.
AUSTEDO XR was FDA approved based on 3 phase 1 studies in healthy volunteers; a steady-state bioequivalence testing study, a dose-proportionality study, and a single-dose relative bioavailability and food effects study.16 The characteristics of each study are shown below:
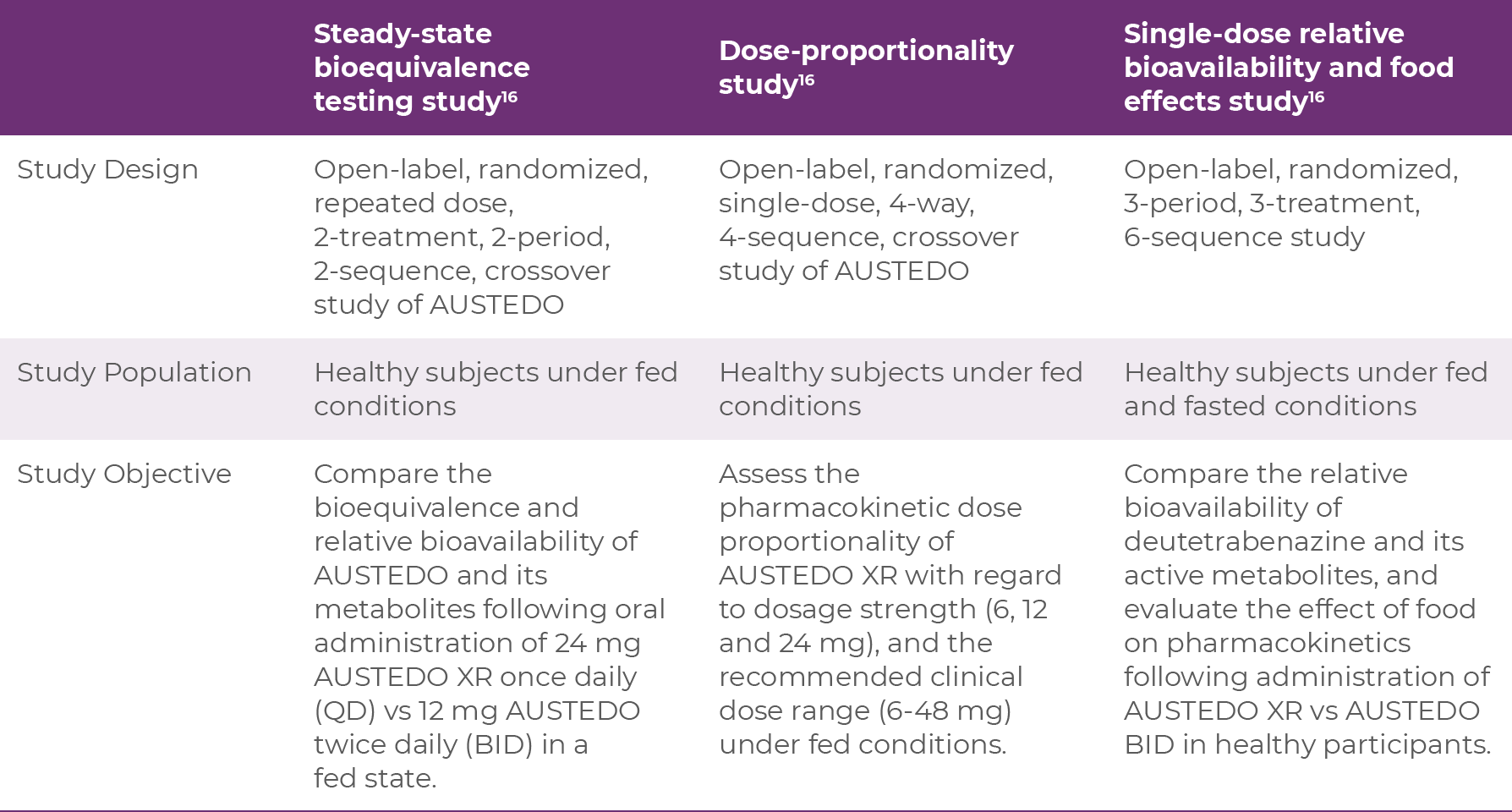
Bioequivalence of once-daily AUSTEDO XR was established with AUSTEDO BID, using pharmacokinetic profile studies. When studies show a drug to be bioequivalent, they can be considered therapeutically equivalent. Data illustrating the bioequivalence of AUSTEDO BID and AUSTEDO XR are shown below (Figure 3.1).16
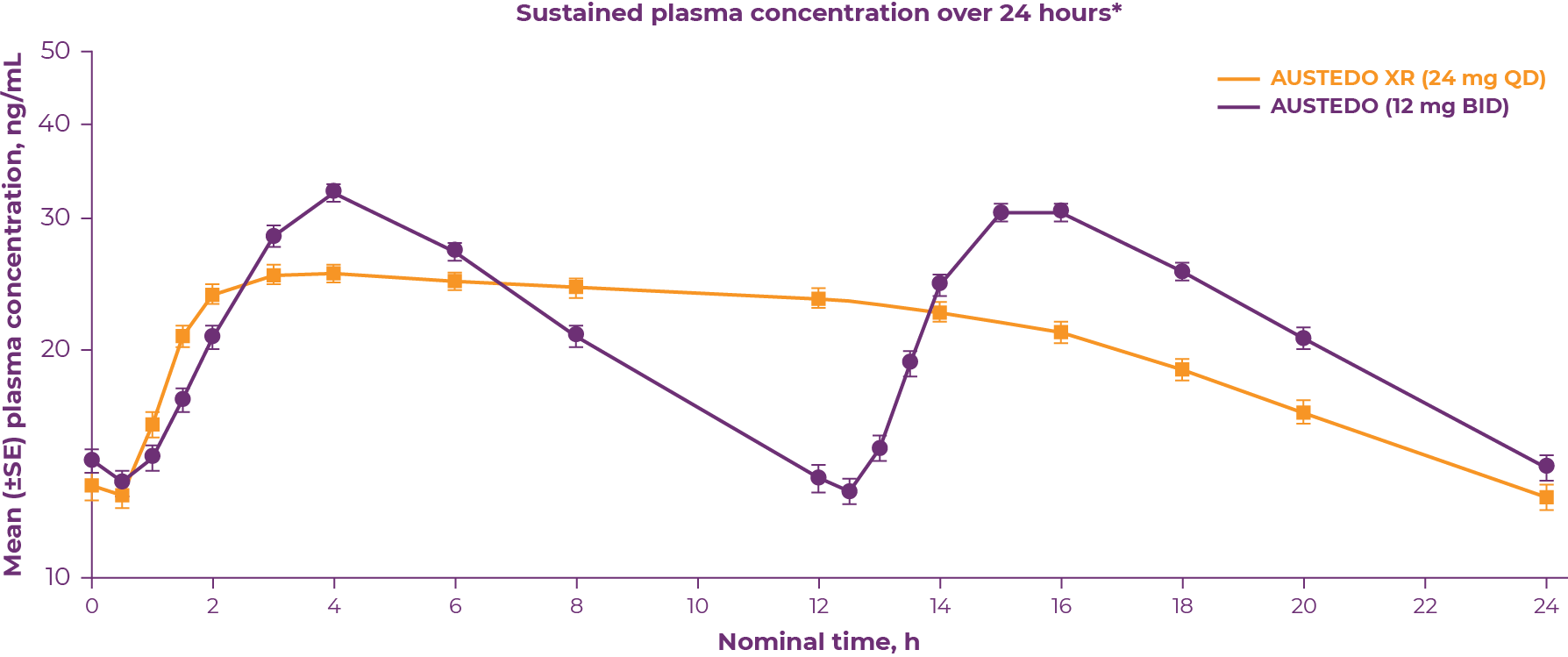 *Bioequivalence was determined by the plasma concentration-time curve from time 0-24 h at steady state of DTBZ (parent) and deuterated α-HTBZ and β-HTBZ metabolites.
*Bioequivalence was determined by the plasma concentration-time curve from time 0-24 h at steady state of DTBZ (parent) and deuterated α-HTBZ and β-HTBZ metabolites.
| • | Bioequivalence of once-daily AUSTEDO XR has been established with AUSTEDO BID based on pharmacokinetic profile studies |
| • | Data support equivalence of AUSTEDO XR and AUSTEDO BID across the full clinical dosing range |
The pivotal trials of AUSTEDO include two randomized, placebo-controlled 12-week clinical trials (Aim to Reduce Movements in Tardive Dyskinesia [ARM-TD] and Addressing Involuntary Movements in Tardive Dyskinesia [AIM-TD]).12-14 Patients who completed ARM-TD or AIM-TD were eligible to enroll in the Reducing Involuntary Movements in Participants With Tardive Dyskinesia (RIM-TD) clinical trial, an open-label study to evaluate long-term maintenance therapy with AUSTEDO for adult patients with TD.17 RIM-TD is the longest TD study to date, with data spanning 688 patient-years through Week 158.16
Patients in the ARM-TD and AIM-TD clinical trials received the AUSTEDO twice-daily (BID) formulation. ARM-TD was a flexible-dose trial in which patients with TD received AUSTEDO titrated to individualized doses that reduced abnormal movements and was tolerated (up to 48 mg/day; mean dose was 38.3 mg/day).12,14 AIM-TD was a fixed-dose trial in which adults with TD were randomized to receive AUSTEDO at doses of 12 mg/day, 24 mg/day, 36 mg/day, or placebo.13,14 The primary endpoint of both studies was the change in Abnormal Involuntary Movement Scale (AIMS) total score from baseline to Week 12.12,13
After completing ARM-TD or AIM-TD, patients in the RIM-TD trial discontinued the study drug for at least 1 week and began AUSTEDO at a dose of 12 mg/day, which was titrated for up to 6 weeks to identify a dose that adequately controlled dyskinesia and was tolerated.17 The dose was adjusted weekly, in increments of 6 mg/day to identify a dose that adequately controlled dyskinesia and was tolerable.17
In the ARM-TD study, AIMS total score for patients receiving AUSTEDO demonstrated statistically significant improvement, by 3.0 points from baseline to endpoint (Week 12), compared with 1.6 points for the placebo group (P=0.019, treatment effect of −1.4 points) (Figure 3.2).12
.png) LS, least squares; SE, standard error.
LS, least squares; SE, standard error.
Patients in the ARM-TD study received the AUSTEDO BID formulation.
In the AIM-TD study, AUSTEDO significantly reduced AIMS total score from baseline to Week 12 by 3.3 points in the 36 mg/day group (P=0.001, treatment difference of −1.9 points), by 3.2 points in the 24 mg/day group (P=0.003, treatment difference of −1.8 points), and by 2.1 points in the 12 mg/day group (P=0.217, treatment difference of −0.7 points) compared with a reduction of 1.4 points in the placebo group (Figure 3.3).13
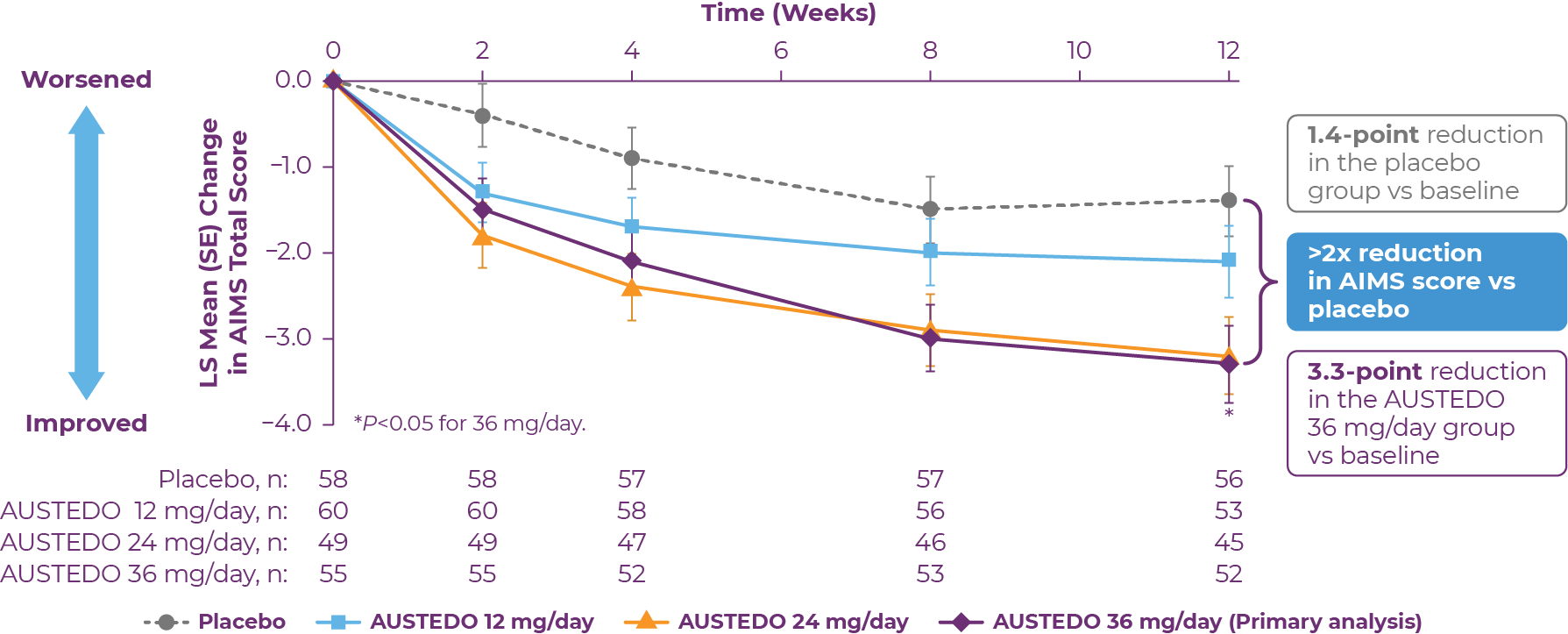 Patients in the AIM-TD study received the AUSTEDO BID formulation.
Patients in the AIM-TD study received the AUSTEDO BID formulation.
In the RIM-TD trial, patients on AUSTEDO experienced a gradual reduction in AIMS total scores over time (Figure 3.4), with 67% and 42% of patients achieving ≥50% and ≥70% improvement in AIMS total score, respectively, by Week 145.17
.png) *Mean total daily dose.
*Mean total daily dose.
Patients in the RIM-TD study received the AUSTEDO BID formulation.
Across both placebo-controlled studies in patients with TD, adverse reactions reported in ≥2% of patients treated with AUSTEDO are shown in Table 3.1.14,18 Across both studies, 4% of patients required dose reduction of AUSTEDO due to adverse events (AEs) versus 2% of patients taking placebo.14 At baseline, 56% of patients treated with AUSTEDO across both studies were taking concomitant antidepressant therapy.18
.png)
Adverse reactions with AUSTEDO XR are expected to be similar to AUSTEDO.
In the RIM-TD trial, tolerability was maintained through Week 158, suggesting that long-term treatment with AUSTEDO has demonstrated tolerability in patients with TD, including those who are concomitantly taking APDs.17
The TD patient population is diverse, comprising patients with myriad comorbid conditions that require various and varied medications.12,13 Polypharmacy and pathways of drug metabolism are important considerations when choosing a treatment for TD, as drug-drug interactions can become a concern for patients taking concomitant medications.14
Clinical trials of AUSTEDO included patients with a broad range of primary conditions, including gastrointestinal disorders, depression, bipolar disorder, schizophrenia, and schizoaffective disorder.12,13,18 Overall, approximately 98% of patients enrolled in AIM-TD and ARM-TD had an underlying psychiatric comorbidity for which they were receiving concomitant medications, including, but not limited to, antidepressants, APDs, anxiolytics, antiepileptics, hypnotics and sedatives, and opioids. Notably, 64% of patients were receiving concomitant atypical APDs and 12% were receiving concomitant typical or combination APDs.14
These patients also had multiple comorbid conditions, including a range of cardiovascular disorders, such as arrythmia, hypertensive heart disease, atrial fibrillation, coronary artery disease, and myocardial ischemia.18 In addition, patients had a history of metabolism and lipid disorders, including diabetes, dyslipidemia, hypercholesterolemia, and hyperlipidemia. Patients were taking a variety of medications to treat these comorbidities, including angiotensin-converting enzyme (ACE) inhibitors, beta blockers, antiarrhythmic drugs, blood-glucose lowering drugs, and lipid-modifying agents.
Approximately half of all medications overall are metabolized through the CYP3A4 pathway.19 Another 30% of medications are metabolized through the CYP2D6 pathway. AUSTEDO XR is metabolized primarily through CYP2D6, with minor contribution from CYP3A4/5 and other enzymes, but AUSTEDO XR is neither an inducer nor inhibitor for these enzyme pathways.14,20 Because AUSTEDO XR is principally metabolized through the CYP2D6 pathway, the maximum daily dose of AUSTEDO XR should not exceed 36 mg in patients taking strong CYP2D6 inducers or in those who are poor CYP2D6 metabolizers (Table 3.2).
 *No trials comparing AUSTEDO XR and Ingrezza® have been conducted.
*No trials comparing AUSTEDO XR and Ingrezza® have been conducted.
AUSTEDO XR is the only FDA-approved VMAT2 inhibitor with no recommendations against concomitant use with CYP3A4 inducers or inhibitors; as such, AUSTEDO XR should be considered for patients with TD who are receiving strong CYP3A4 inducers or inhibitors for underlying conditions (Table 3.2).14,20.21
AUSTEDO XR is a VMAT2 inhibitor indicated for the treatment of adult patients with TD.14 AUSTEDO XR has no recommendations against concomitant use with CYP3A4 inhibitors or inducers, and therefore, should be considered for patients taking those medications.
1. Caroff SN, Campbell EC, Carroll B. Pharmacological treatment of tardive dyskinesia: recent developments. Expert Rev Neurother. 2017;17(9):871-881. 2. Caroff SN, Ungvari GS, Cunningham Owens DG. Historical perspectives on tardive dyskinesia. J Neurol Sci. 2018;389:4-9. 3. Alexander GC, Gallagher SA, Mascola A, Moloney RM, Stafford RS. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184. 4. Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet Mov (N Y). 2013;3. 5. Cutler AJ, Caroff SN, Tanner CM, et al. Presence and impact of possible tardive dyskinesia in patients prescribed antipsychotics: results from the RE-KINECT study. CNS Spectr. 2019;24(1):176-177. 6. Caroff SN. Overcoming barriers to effective management of tardive dyskinesia. Neuropsychiatr Dis Treat. 2019;15:785-794. 7. Ascher-Svanum H, Zhu B, Faries D, Peng X, Kinon BJ, Tohen M. Tardive dyskinesia and the 3-year course of schizophrenia: results from a large, prospective, naturalistic study. J Clin Psychiatry. 2008;69(10):1580-1588. 8. Lieberman JA, Alvir J, Geisler S, et al. Methylphenidate response, psychopathology and tardive dyskinesia as predictors of relapse in schizophrenia. Neuropsychopharmacology. 1994;11(2):107-118. 9. McEvoy J, Park T, Schilling T, Terasawa E, Ayyagari R, Carroll B. The burden of tardive dyskinesia secondary to antipsychotic medication use among patients with mental disorders. Curr Med Res Opin. 2019;35(7):1205-1214. 10. Caroff SN, Davis VG, Miller DD, et al; CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303. 11. Keepers GA, Fochtmann LJ, Anzia JM, et al. The American Psychiatric Association Practice Guideline for the Treatment of Patients With Schizophrenia. 3rd ed. American Psychiatric Association; 2021. 12. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010. 13. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604. 14. AUSTEDO® XR (deutetrabenazine) extended-release tablets/AUSTEDO® current Prescribing Information. Parsippany, NJ: Teva Neuroscience, Inc. 15. Scorr LM, Factor SA. VMAT2 inhibitors for the treatment of tardive dyskinesia. J Neurol Sci. 2018;389:43-47. 16. Hauser RA, Barkay H, Fernandez HH, et al. Long-term deutetrabenazine treatment for tardive dyskinesia is associated with sustained benefits and safety: a 3-year, open-label extension study. Front Neurol. 2022;13:773999. 17. Data on file. Teva Neuroscience, Inc. 18. Fernandez HH, Stamler D, Davis MD, et al. Long-term safety and efficacy of deutetrabenazine for the treatment of tardive dyskinesia. J Neurol Neurosurg Psychiatry. 2019;90(12):1317-1323. 19. Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48(11):689-723. 20. Center for Drug Evaluation and Research. AUSTEDO Clinical Pharmacology and Biopharmaceutics Review. Application number: 208082Orig1s000. February 27, 2017. Accessed May 3, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208082Orig1s000ClinPharmR.pdf 21. Ingrezza® (valbenazine) capsules. Prescribing Information. San Diego, CA: Neurocrine Biosciences, Inc.
IMPORTANT SAFETY INFORMATION
Indications and Usage
AUSTEDO® XR (deutetrabenazine) extended-release tablets and AUSTEDO® (deutetrabenazine) tablets are indicated in adults for the treatment of chorea associated with Huntington's disease and for the treatment of tardive dyskinesia.
Important Safety Information
Depression and Suicidality in Patients with Huntington's Disease: AUSTEDO XR and AUSTEDO can increase the risk of depression and suicidal thoughts and behavior (suicidality) in patients with Huntington's disease. Balance the risks of depression and suicidality with the clinical need for treatment of chorea. Closely monitor patients for the emergence or worsening of depression, suicidality, or unusual changes in behavior. Inform patients, their caregivers, and families of the risk of depression and suicidality and instruct them to report behaviors of concern promptly to the treating physician. Exercise caution when treating patients with a history of depression or prior suicide attempts or ideation. AUSTEDO XR and AUSTEDO are contraindicated in patients who are suicidal, and in patients with untreated or inadequately treated depression.
Contraindications: AUSTEDO XR and AUSTEDO are contraindicated in patients with Huntington's disease who are suicidal, or have untreated or inadequately treated depression. AUSTEDO XR and AUSTEDO are also contraindicated in: patients with hepatic impairment; patients taking reserpine or within 20 days of discontinuing reserpine; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; and patients taking tetrabenazine or valbenazine.
Clinical Worsening and Adverse Events in Patients with Huntington's Disease: AUSTEDO XR and AUSTEDO may cause a worsening in mood, cognition, rigidity, and functional capacity. Prescribers should periodically re-evaluate the need for AUSTEDO XR or AUSTEDO in their patients by assessing the effect on chorea and possible adverse effects.
QTc Prolongation: AUSTEDO XR and AUSTEDO may prolong the QT interval, but the degree of QT prolongation is not clinically significant when AUSTEDO XR or AUSTEDO is administered within the recommended dosage range. AUSTEDO XR and AUSTEDO should be avoided in patients with congenital long QT syndrome and in patients with a history of cardiac arrhythmias.
Neuroleptic Malignant Syndrome (NMS), a potentially fatal symptom complex reported in association with drugs that reduce dopaminergic transmission, has been observed in patients receiving tetrabenazine. The risk may be increased by concomitant use of dopamine antagonists or antipsychotics. The management of NMS should include immediate discontinuation of AUSTEDO XR and AUSTEDO; intensive symptomatic treatment and medical monitoring; and treatment of any concomitant serious medical problems.
Akathisia, Agitation, and Restlessness: AUSTEDO XR and AUSTEDO may increase the risk of akathisia, agitation, and restlessness. The risk of akathisia may be increased by concomitant use of dopamine antagonists or antipsychotics. If a patient develops akathisia, the AUSTEDO XR or AUSTEDO dose should be reduced; some patients may require discontinuation of therapy.
Parkinsonism: AUSTEDO XR and AUSTEDO may cause parkinsonism in patients with Huntington's disease or tardive dyskinesia. Parkinsonism has also been observed with other VMAT2 inhibitors. The risk of parkinsonism may be increased by concomitant use of dopamine antagonists or antipsychotics. If a patient develops parkinsonism, the AUSTEDO XR or AUSTEDO dose should be reduced; some patients may require discontinuation of therapy.
Sedation and Somnolence: Sedation is a common dose-limiting adverse reaction of AUSTEDO XR and AUSTEDO. Patients should not perform activities requiring mental alertness, such as operating a motor vehicle or hazardous machinery, until they are on a maintenance dose of AUSTEDO XR or AUSTEDO and know how the drug affects them. Concomitant use of alcohol or other sedating drugs may have additive effects and worsen sedation and somnolence.
Hyperprolactinemia: Tetrabenazine elevates serum prolactin concentrations in humans. If there is a clinical suspicion of symptomatic hyperprolactinemia, appropriate laboratory testing should be done and consideration should be given to discontinuation of AUSTEDO XR and AUSTEDO.
Binding to Melanin-Containing Tissues: Deutetrabenazine or its metabolites bind to melanin-containing tissues and could accumulate in these tissues over time. Prescribers should be aware of the possibility of long-term ophthalmologic effects.
Common Adverse Reactions: The most common adverse reactions for AUSTEDO (>8% and greater than placebo) in a controlled clinical study in patients with Huntington's disease were somnolence, diarrhea, dry mouth, and fatigue. The most common adverse reactions for AUSTEDO (4% and greater than placebo) in controlled clinical studies in patients with tardive dyskinesia were nasopharyngitis and insomnia. Adverse reactions with AUSTEDO XR extended-release tablets are expected to be similar to AUSTEDO tablets.
Please see accompanying full Prescribing Information, including Boxed Warning.

Austin, Texas
Dr Rakesh Jain considers multiple factors contributing to drug-drug interactions (DDIs) and recounts his approach to managing multiple medications in patients using antipsychotic medications.
Q:What proportion of your psychiatric/tardive dyskinesia (TD) patients are suffering with comorbidities that require chronic medication? Which conditions are most frequent?
A:It is very common; 90% or more of patients have multiple psychiatric diagnoses. For instance, in bipolar disorder, a patient may be taking up to 6 psychiatric medications at the same time. Anxiety and insomnia are very common comorbidities, then there are also mood disorders, attention-deficit hyperactivity disorder, and impulse control disorders. For insomnia alone, concomitant medications can include benzodiazepines, anticholinergics, and melatonin, and don't forget that patients may self-medicate with alcohol. Nonpsychiatric comorbidities are also frequent; greater than 50% of patients have comorbid conditions, and the likelihood corresponds with age. Obesity almost always creeps in by age 50, along with the resultant metabolic dysregulations and heart conditions. Cardiac medications are quite common in the psychiatric patient population, and there are several drug classes liable to DDIs, such as calcium channel blockers, antiarrhythmic drugs, and lipid lowering agents.
Q:What are your primary concerns around drug-drug interactions when prescribing a drug for one of your psychiatric patients with TD?
A:I am concerned about the usual issues: safety events, poor tolerability, and loss of efficacy. Excessive activity is another key concern; our medications live on a relatively modest spectrum of safety. Even with selective serotonin reuptake inhibitors (SSRIs), if you overdo it, the risk of serotonin syndrome is considerable. Excessive serotonin and norepinephrine reuptake inhibitor (SNRI) activity could give the patient hypertension. If the blood levels go up too much for these drugs, it can destabilize everything, and the whole house of cards collapses. You might have to pull away every medication and start over again. This strongly supports the notion that before you embark on a journey, you should know what the potential dangers are; and drug-drug interactions are most certainly one of those dangers. I have to be cognizant of the possible interactions before I prescribe something new.
Psychiatrists also need to be aware of how other healthcare providers (HCPs) are managing comorbid conditions with medication. It's like in a parking lot where two cars are backing up and not aware of each other; neither one has bad intentions, but boom, now you've got a fender bender. I could end up causing a cardiac instability for a patient, perhaps their arrhythmia control or hypertension, so the consequences are significant and multiple. It is a better approach to know the landscape, pharmacokinetically, before you embark on this journey.
Q:Upon prescribing a new medication, how do you assess your patients for emergence of a problem?
A:It's a combination of proactive inquiry from my office and patient-initiated report, and it depends on the patient. Some of them, you can count on to inform you of a challenge that they experience, but other patients may simply stop taking their medications rather than informing you. And once patients experience a side effect while taking a new drug, they may never feel comfortable about taking it again. Psychiatrists sometimes prefer to begin a new drug at a subtherapeutic dose to avoid side effects, but this has its own downside: sometimes the patient feels no benefit from the new drug and decides to stop taking it. It is a much better approach for drug selection to be guided by possible DDIs; this allows you to advance more confidently to therapeutic concentrations.
Q:Cytochrome P3A4 (CYP3A4) is a key metabolic pathway for many commonly used drugs. What is your process for integrating multiple medications in your psychiatric patients with TD when a CYP3A4 drug is on board?
A:Yes, it's a MAJOR pathway, a superhighway. One mistake psychiatry does make is to confuse the different modes of interaction with the P450 enzyme pathways. It is key to distinguish a substrate: a drug acted on by the P450 enzyme, from an inducer or inhibitor, which affects the activity of the enzyme on other drugs. The US Food and Drug Administration (FDA) terminology is that the drug that causes a change in metabolic activity is the "perpetrator drug," and the drug with affected plasma levels and clearance is the "victim drug." Frequently, the perpetrator drug is also a substrate for the same enzyme pathway it affects. Just knowing "that's a 3A4 drug" doesn't give enough information to be useful or to guide decision-making. For instance, a substrate drug, the victim, may have alternative routes of metabolism, which is good, because it gives a little more flexibility for use with a perpetrator drug. But some substrate drugs don't have other routes of metabolism or clearance, and that can be a big deal!
Among the perpetrator drugs, drugs that are inducers or inhibitors, the distinction between weak, moderate, or strong inhibitor or inducer is very important. Rarely do warnings apply to weak perpetrators. The FDA contraindicates the use of many psychiatric drugs if a strong or moderate CYP3A4 inhibitor or inducer is also prescribed; for instance, lurasidone cannot be used with a strong 3A4 inducer or inhibitor. Alternatively, FDA may delineate in the product label what can and can't be done for victim drugs in the presence of a perpetrator, such as dose modification. And don't forget the impact of DDIs on adherence to the antipsychotic medication. Poor adherence can really destabilize patients' mental health.
Lastly, when multiple drugs sharing metabolic pathways are coadministered and a problem emerges, attribution can be quite challenging, owing to the dimension of time. It can take 2 weeks for the effects of an enzyme inducer to maximally affect enzyme activity, and a similar length of time to return to baseline activity after drug withdrawal.
Q:CYP2D6 is less frequently implicated in DDIs. What is your level of concern with CYP2D6, and how do you handle the risk in your patients?
A:This is another important oxidative metabolic pathway in psychiatry, although a slightly lower proportion of drugs go through this pathway. The same DDI concerns exist with CYP2D6, plus there can be intrinsically different levels of CYP2D6 activity in patients due to genetic variability of the enzyme itself.
Q:Since genetic variation in cytochrome P450 enzyme activity can also affect drug levels, leading to safety or efficacy issues, how do you accommodate this possible source of variance in your medication management?
A:Currently, there is not a high likelihood that prescribers would know the pharmacogenomic profile of one of their patients. And when you think about it, there is not that big an advantage in knowing. Even if I have an extensive metabolizer, at the drop of a hat, we can make that patient a functional "poor metabolizer" with a drug. So, it's not a static status; the patient's status can be altered within 3 or 4 days just by taking a new drug, or dietary supplement, or even a grapefruit juice.
Genotyping is a step in the right direction, but the biggest bang for the buck is knowing what medications your patient is on, and how those drugs impact oxidative pathways.
Q:Interactions based on drug metabolism pathways are well understood, but what are your thoughts on drug transporters, such as P-glycoprotein, as drivers of adverse interactions? How aware are HCPs of possible transporter DDI?
A:There is a very low awareness and understanding of the impact of drug transporter proteins on DDIs, and it must be acknowledged that there is limited guidance available in this space as well. There are not that many drugs for which we have actionable clinical information on drug transporters, but it's an area that many people are looking at, including the FDA.
Q:What is the role of AUSTEDO XR in managing TD for patients with concomitant medications?
A:AUSTEDO XR is an appropriate choice when patients could have a multitude of drugs with CYP3A4 issues. For instance, let's say I have a 75-year-old patient with a cardiac history, and today all the drug-drug combinations look clean. But I am fully expecting that there will be more changes to the medication regimen due to the patient's age and history; it's just going to happen. I might consider using deutetrabenazine in anticipation of avoiding DDIs down the road. It's a medication worthy of thought, but not the only choice for all cases, and watching the patient for emergent problems is still necessary.
Q:What are some things that HCPs can do to mitigate the possibility of pharmacokinetic/metabolism issues emerging in their psychiatric/TD patients because of DDI? What are best practices for working with other HCPs who care for the same patient?
A:Maintaining awareness is such an important facet, and we need to be doing a better job. Some psychiatric patients don't get good care to begin with, and, frequently, the primary care physician focuses narrowly on the conditions and drugs that they are managing, but it's the same patient body! And we can't count on the patients to know all the medications they are on, the metabolic pathways and areas of concern. As a patient myself, I don't know the metabolic pathways of medications I'm on, I expect my clinician to know!
The art of prescribing requires us to know both the pharmacodynamics and the pharmacokinetics. We must appreciate that 50% of drug prescribing is pharmacokinetics and that it's a marriage of equals. We can't divide our thinking between pharmacodynamics and pharmacokinetics, divide no more, because it may put our patients at risk.
Lastly, we need to be aware and recognize that there will be scenarios of DDIs in many of our patients. The key is to know the metabolic pathways that could be involved so you can quickly understand where the problem exists and manage it in a focused way.
Barnes TR, Bhatti SF, Adroer R, Paton C. Screening for the metabolic side effects of antipsychotic medication: findings of a 6-year quality improvement programme in the UK. BMJ Open. 2015;5(10):e007633.
Bleakley S. Antidepressant drug interactions: evidence and clinical significance. Prog Neurol Psychiatry. 2016;20(3):21-27. https://wchh.onlinelibrary.wiley.com/doi/10.1002/pnp.429
Horn JR, Hansten PD. Hansten & Horn Drug Interactions: Get to know an enzyme: CYP2D6. Pharmacy Times. 2008;0:0. Accessed August 12, 2020. https://www.pharmacytimes.com/view/2008-07-8624
Horn JR, Hansten PD. Hansten & Horn Drug Interactions: Get to know an enzyme: CYP3A4. Pharmacy Times. 2008;0:0. Accessed August 2, 2020. https://www.pharmacytimes.com/view/2008-09-8687
Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H. Inhibition and induction of human cytochrome P450 enzymes: current status. Arch Toxicol. 2008;82(10):667-715.
Saravanakumar A, Sadighi A, Ryu R, Akhlaghi F. Physicochemical properties, biotransformation, and transport pathways of established and newly approved medications: a systematic review of the top 200 most prescribed drugs vs. the FDA-approved drugs between 2005 and 2016. Clin Pharmacokinet. 2019;58(10):1281-1294.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions: Guidance for Industry. Docket number FDA-2017-D-5961. Accessed April 21, 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions
Weinstock LM, Gaudiano BA, Epstein-Lubow G, Tezanos K, Celis-Dehoyos CE, Miller IW. Medication burden in bipolar disorder: a chart review of patients at psychiatric hospital admission. Psychiatry Res. 2014;216(1):24-30.
Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48(11):689-723.